Development stage
- Lead Identification
- In Vivo Efficacy
- PK & Metabolism
- Toxicity
- Chemistry & Manufacturing
Health Condition & Disease
Breast cancer is the most commonly diagnosed cancer among women and the second leading cause of cancer death in women. According to American Cancer Society, there are 281,550 new cases of invasive breast cancer each year, 49,290 new cases of non-invasive (in situ) breast cancer, and 43,600 deaths caused by breast cancer. Breast cancer is a complex and heterogeneous disease with a wide variation in clinical responses, which complicates the identification of the best treatment option. Among different subtypes of breast cancer, triple negative breast cancer (TNBC) is particularly difficult to treat and has the worst prognosis because of lacking expression of estrogen receptor (ERa), progesterone receptor (PR), and HER2. Due to the absence of a recognizable therapeutic target, the systemic treatment options for TNBC are limited to cytotoxic chemotherapy, which is used before and after surgery and for advanced cancer that has spread outside the breast and underarm area. DNA-targeting agents like cyclophosphamide, fluorouracil, and gemcitabine are some of the most widely used TNBC drugs. Although chemotherapy is effective in some patients, its long-term outcome is poor due to the severe side effects. Thus, novel strategies are urgently needed to augment the efficacy and selectivity of chemotherapeutic agents for TNBC treatment.
Drug Lead Identification
To address this need, the Peng group has developed reactive oxygen species (ROS)-activated DNA alkylating agents that exhibit selective toxicity towards breast cancer cells, which have inherently high levels of ROS. An electron-withdrawing H2O2-responsive boronate group was introduced to mask alkylating moiety, while H2O2 can transform the boronate group to a donating OH functionality to restore the reactivity of DNA alkylators. Dr. Gandhi (MD Anderson Cancer Center) group demonstrated that the ROS-activated DNA alkylating agents are not toxic to normal cells isolated from healthy donors at the doses needed to kill cancer cells. Dr. Peng group has worked together with Dr. Arnold to establish proof-of-concept in meeting critical design features for a new drug targeting TNBC cells, including increased in vivo efficacy and high selectivity for TNBC, lack of toxicity, microsomal stability, and good pharmacokinetic properties. Most of these ROS-activated compounds are non-toxic but can be selectively activated by high-level of H2O2 to release DNA cross-linkers, either quinone methide or nitrogen mustards that effectively alkylate DNA. Compound 1 served as lead compound for further optimization and structure activity relationship study which resulted in pharmacological improvements to achieve critical design features (Figure 1).
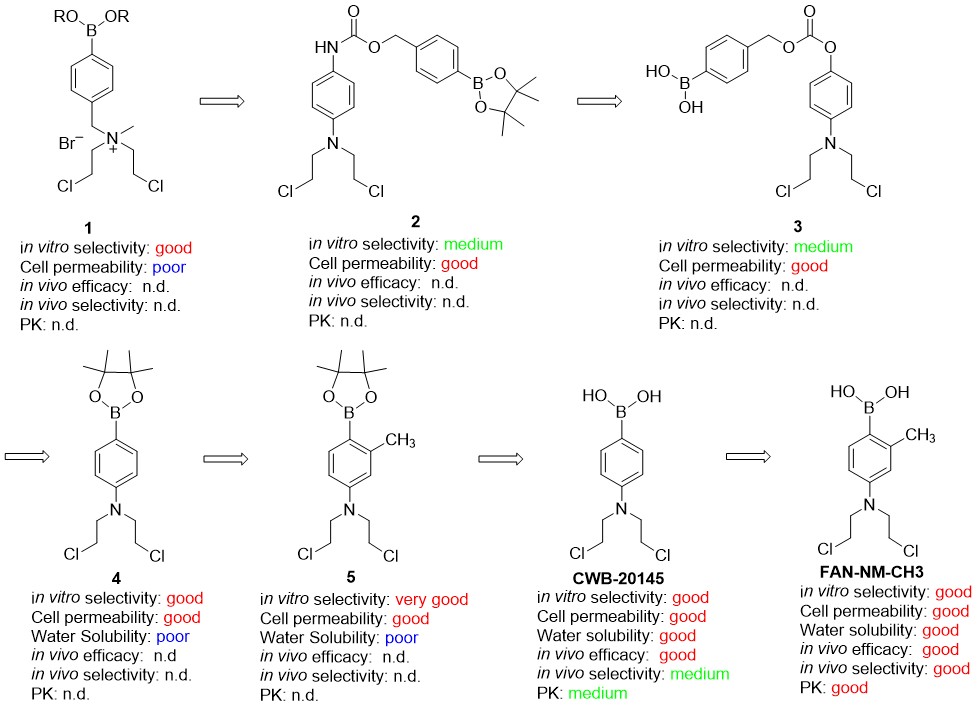
Since the reactivity of nitrogen mustard strongly depends on the electron density on nitrogen, there are three ways to shield its reactivity, including introducing a positive charge on mustard nitrogen (1), an electron withdrawing linker (2 and 3), or directly attaching the boronate group to an aromatic nitrogen mustard (4 and 5) (Figure 1). Compound 1 showed selective toxicity towards various cancer cells in contrast to normal cells. However, the cellular activity of 1 is low due to insufficient permeability caused by the positively charged nitrogen, which is necessary to deactivate the nitrogen mustard. To address this issue, two classes of neutral prodrugs have been designed and synthesized, which displayed enhanced permeability and increased cellular activity and selectivity. Compound 3 induced over 50% growth inhibition of numerous cancer cell lines, including: leukemia, non-small cell lung cancer (NSCLC), breast cancer and renal cancer with a GI50 of less than 1 μM. Other analogs, such as 4 and 5, containing a boronate group directly attached to the benzene ring, showed superior anticancer activity and specificity than previous ROS-activated nitrogen mustard prodrugs. Among those agents, boronic acid analogs CWB-20145 and Fan-NM-CH3 are the most promising ROS-activated nitrogen mustard prodrugs that showed in vivo efficacy and selectivity with improved water solubility. CWB-20145 presents a reduced toxicity than chlorambucil yet retains potent antitumor effects. Furthermore, its methyl analog Fan-NM-CH3 further improved in vivo efficacy and tumor specificity as well as pharmacokinetic properties.
In vivo efficacy and selectivity
The currently lead compound FAN-NM-CH3 retained all properties of CWB-20145 with improved PK properties. FAN-NM-CH3 is 10 times more cytotoxic against TNBC cell line than chlorambucil and 16 times more active than melphalan. In comparison with CWB-20145 that showed rapid conversion, FAN-NM-CH3 with a methyl group greatly increased in vivo duration and absorption. In vivo toxicity study in mice demonstrated that both CWB-20145 and FAN-NM-CH3 were less toxic than chlorambucil. Both compounds reduced tumor growth with an inhibition rate more than 90% in athymic nude mice xenografted with TNBC MDA-MB-468 cells. The in vivo investigation also indicated that the methyl analogue FAN-NM-CH3 showed a better safety profile than the parent compound CWB-20145.
Pharmacokinetics and Metabolism
In the presence of human liver microsome, CWB-20145 and FAN-NM-CH3 showed a t1/2 of 60 min and 84 min, respectively. The blood concentrations of CWB-20145 and FAN-NM-CH3 were readily detected in plasma with a tmax of about three min. The presence of a methyl group in FAN-NM-CH3 increased the half-life to 9 min from 5 min for CWB-20145. The rate of activation for CWB-20145 in the blood was fast (0.141 min−1), which is two times faster than that of FAN-NM-CH3 (0.078 min−1). Compound FAN-NM-CH3 showed an area under the curve (AUC) of 16253 ng•min/mL, which is higher than that of CWB-20145 (10883 ng•min/mL). The determination of metabolites is undergoing.
Safety
The methyl analogue FAN-NM-CH3 showed a better safety profile than the parent compound CWB-20145 and the clinically used DNA alkylating agents, such as chlorambucil. Single dose treated mice survived at a maximal tolerated dose of 80 mg/kg CWB-20145 and 100 mg/kg FAN-NM-CH3. Chlorambucil, however, induced death at 80 mg/kg for all animals. A repeated-dose toxicity study indicated that chlorambucil induced death at 40 mg/kg repeated dose on day 3, while all mice treated daily with 50 mg/kg 1 or 2 survived. Furthermore, FAN-NM-CH3-treated xenografted mice did not show loss of appetite, reduced activity levels, or color/weight changes for lung, liver, spleen, heart, and kidney after 6 week IP administration of FAN-NM-CH3 at a dosage of 5.0 mg/Kg. Slight weight loss was observed for CWB-20145 at the last day of administration, while the FAN-NM-CH3-treated mice showed a similar increase in body weight as compared to the control group. By contrast, the IP administration of chlorambucil (5.0 mg/Kg) resulted in severe toxicity with more than 15% body weight loss.
Chemistry and Manufacturing
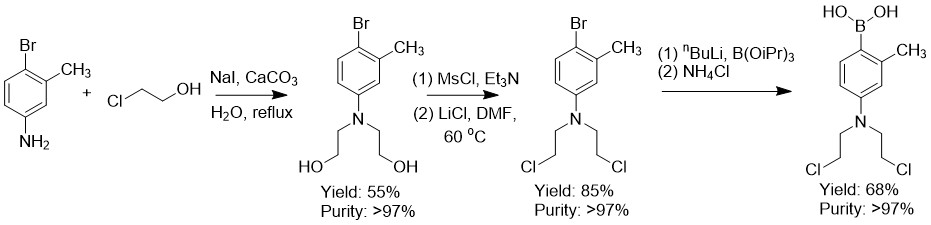
FAN-NM-CH3 was synthesized starting from commercially available 4-bromo-2-methylaniline in a total yield of 32%. All intermediates were isolated with a purity of >97%. A 5 g batch of FAN-NM-CH3 was isolated in a 32% overall yield and a purity of >97% after precipitation.