Development stage
- Lead Identification
- In Vivo Efficacy
- PK & Metabolism
- Toxicity
- Chemistry & Manufacturing
Health Condition & Disease

Asthma is a major global health concern, occurring in more than 300 million persons worldwide, with 26.5 million affected individuals in the US alone. A hallmark of asthma is chronic airway inflammation, leading to airway remodeling, hyperactivity to external stimuli, and airway obstruction. Yet, current therapeutic options to control symptoms are limited. Persistent asthma is treated with first-line inhaled corticosteroids (ICS) that are supplemented in more advanced disease with long-acting β2 adrenergic receptor agonists (LABA), oral corticosteroids, or other immune modulating agents. Leukotriene receptor antagonists are oral alternatives for patients with disease poorly controlled with inhaled steroids, however, genetic variations in leukotriene signaling genes may preclude efficacy in a large number of patients. Injectable biologics that target Th2 mediated asthmatic eosinophilia have been approved, but high treatment costs limit their use to only severe disease. Taken together, effective asthma treatment represents an unmet medical need for a large number of patients whose disease is not adequately controlled with current therapeutic options.
Drug Lead Identification
To address this need, the Arnold Group has worked together with the research group of Dr. Emala (Columbia University), who identified the expression of gamma amino butyric acid type A receptors (GABAAR) on airway smooth muscle cells and demonstrated that GABAAR ligands relax airway smooth muscle though inhibition of calcium handling. More than a hundred GABAAR ligands (imidazobenzodiazepines) were synthesized by the Cook Group and evaluated by the Arnold Group to establish proof-of-concept in meeting critical design features for a new asthma drug that targets GABAARs, including: oral bioavailability, microsomal stability and good PK, high lung distribution, low CNS exposure, lack of toxicity, and effectiveness in reducing airway hyperresponsiveness (AHR) and inflammation. All compounds studied are based on two different scaffolds that bind preferentially to α4 or α5 containing GABAAR subtypes. Compound optimization starting with XHE-III-74 laid the foundation for a preliminary structure activity relationship scheme resulting in key pharmacological improvements to achieve the critical design features (Figure 1).
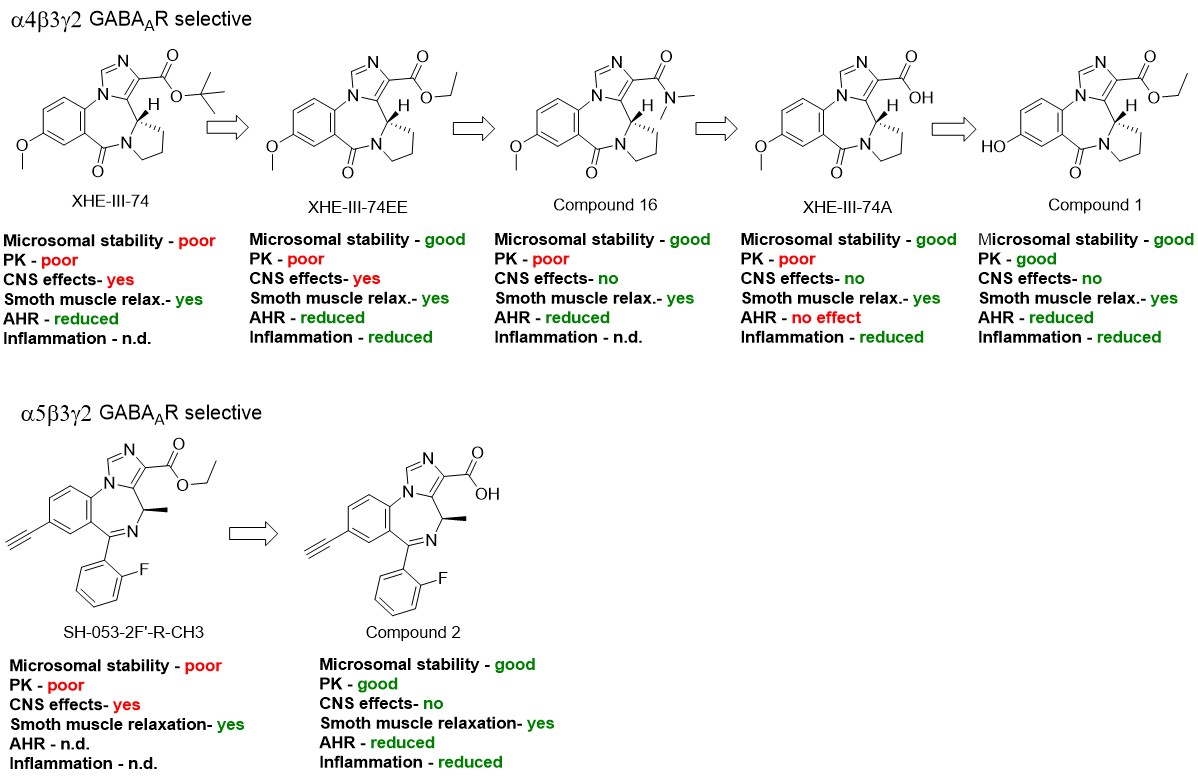
Initially, a series of α4 selective compounds with different esters were investigated resulting in XHE-III-74EE, which showed improved microsomal stability in human and mouse liver microsomes. This pharmacological property is essential for translation of in vivo mouse disease models to human drug development. Next, a series of different amides were investigated, which resulted in compound 16 that significantly reduced distribution across the mouse blood brain barrier (BBB). Avoidance of brain exposure is a key asthma drug design feature to preclude any activity at GABAAR expressed on neurons that could lead to CNS adverse effects. A further improvement was achieved with XHE-III-74A, where the acid functionality blocked BBB transit entirely. Further development led to compound 1 demonstrating excellent oral availability and a four-hour half-life in lung with negligible amounts measurable in brain. Thus, the phenolic function improved oral availability while inhibiting BBB transit. Similar to XHE-III-74EE, compound 1 was stable in the presence of mouse liver microsomes. Compound 1 reduced the number of inflammatory cells in the asthmatic mouse lung and reduced the transmembrane current of CD4+ T cells. In an ovalbumin sensitized/challenged (ova s/c) asthma model, this compound also reduced methacholine induced AHR. SH-053-2F’-R-CH3, an α5 selective compound, induced relaxation of pre-contracted GP and human tracheal airway smooth muscle (ASM) ex vivo. However, significant brain distribution and rapid metabolism/clearance prompted us to apply knowledge gained in the α4 series to improve compound performance. The corresponding acid, compound 2, was significantly more stable in the presence of mouse and human liver microsomes and did not cross the BBB. In mice, 2 had a long half-life and AHR was reduced significantly in the ova s/c model. Although 2 at 100 mg/kg b.i.d. for five days reduced total inflammatory cell numbers in mice, it did not reduce CD4+ T cells nor reduce their transmembrane current.
Patent
The compounds have been patent as WO2018035246A1 and US9879020B2 in collaboration with the UWM Research Foundation. For detailed information see technology 1319.
In vivo efficacy
The current lead compound MIDD0301 retained all properties of 2 and reduced AHR in the ova s/c model and exhibited significant anti-inflammatory properties. In the ova s/c model, CD4+ T cell numbers were reduced at 20 mg/kg b.i.d. over a period of five days and eosinophils and alveolar macrophages were reduced at 100 mg/kg with the same dosing schedule. IL-17a, TNFα, and IL-4 levels were decreased in the treated asthmatic lung. Changes of the airway mucus layer was not observed. MIDD0301 also relaxed human airway smooth muscle and reduced AHR in a murine house dust mite (HDM) model when nebulized. Later studies have shown that nebulized MIDD0301 exhibited comparable or better therapeutic potency compared to nebulized albuterol. Prophylactic nebulized MIDD0301 was also effective in reducing bronchoconstriction, comparable to nebulized albuterol or fluticasone, in a steroid resistant asthma mouse model. Oral dexamethasone was ineffective in this model. Nebulized MIDD0301 was also effective in reversing bronchospasm when dosed after methacholine challenge comparable to albuterol.
Pharmacokinetics and Metabolism
In the presence of liver and kidney microsomes MIDD0301 is stable for >2 hours but is converted to the corresponding glucuronide and glucoside in the presence of conjugation cofactors. MIDD0301 together with significant amounts of MIDD0301 glucoside and MIDD0301 taurine were observed in urine and feces for IP and IV administration. For oral administration, MIDD0301 glucuronide was identified as the main metabolite. Pharmacokinetic analysis of IV, IP and oral administration showed high concentrations of MIDD0301 in lung and blood. Very low levels of MIDD0301 were found in the brain. The half-lives in these tissues ranged between 4-6 hours for IP and oral and 1-2 hours for IV administration. Nebulized MIDD0301 maintained therapeutics level in the lung for at least 25 minutes. Significant lower concentrations were observed in the blood and brain.
Safety
MIDD0301 was designed to have limited brain distribution resulting in the absence of CNS effects at single oral dose of 1000 mg/kg or when given at 200 mg/kg daily for 28 days. Unlike other imidazodiazepines, MIDD0301 does not accumulate in the brain during that time. To address an immunosuppressive potential of MIDD0301, a 28-day repeat dose immunotoxicity evaluation of MIDD0301 (100 mg/kg, b.i.d.) did not reveal signs of general toxicity as determined by animal weight, organ weight, or hematology. In contrast, 5 mg/kg prednisone-treated mice gained less weight over the course of the study and exhibited reduced spleen and thymus weights. Unlike, prednisone, MIDD0301 did not change circulating lymphocyte, monocyte, and granulocyte numbers and did not alter IgG antibody responses to DNP following DNP-KLH immunization, indicating that systemic humoral immune function was not affected by MIDD0301. Furthermore, MIDD0301 does not inhibit the hERG channel and has shown no adverse cardiovascular effects following 100 mg/kg IP dosing. Initial genotoxicity studies using MIDD0301 (100 µM) with or without cytocalasin B showed similar percent of mononuclear TK6 cells compared to negative control compound acetylsalicylic acid. Investigations of chromosomes of MIDD0301 treated CHO cells did not reveal any DNA break or inappropriate rejoining when stained with Giemsa stain.
Chemistry and Manufacturing
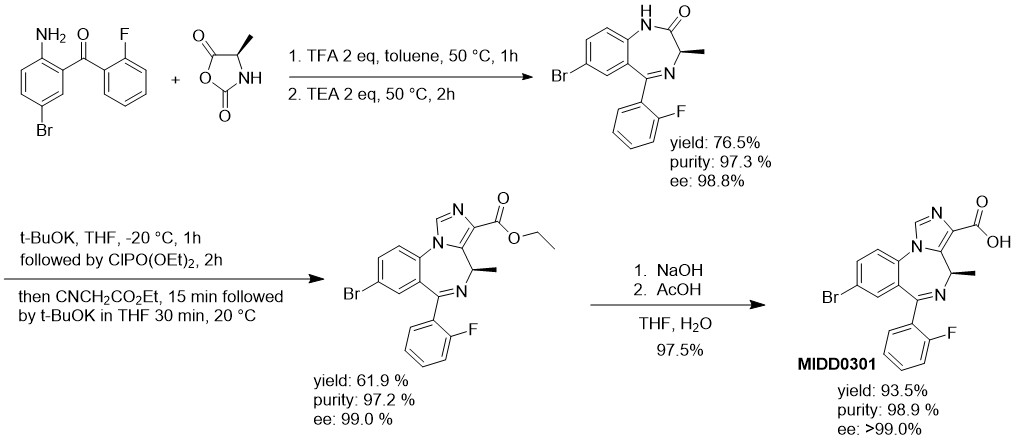
Large scale manufactory difficulties were overcome with the application of the N-carboxyanhydride of d-alanine. Activated in the presence of an acid, this NCA reacted with nonbasic 2-amino-5-bromo-2′-fluorobenzophenone and formed the 1,4-diazepine upon neutralization with triethylamine. Carefully designed workup procedures eliminated the need for column chromatography. Further improvement with large-scale reactors included a temperature-controlled slow addition of reagents to generate the imidazodiazepine at −20 °C. Intermediates were isolated with a purity of >97% and included the characterization of impurity profiles. A 100 g batch of MIDD0301 was isolated in a 44% overall yield and a purity of 98.9% after recrystallization with an optical purity >99.0%.