Development stage
- Lead Identificaiton
- In Vitro Efficacy
Health Condition & Disease
In the past, antiviral drug discovery focused almost exclusively on viruses that cause chronic, life-long, diseases, like AIDS, herpes and hepatitis. The COVID-19 pandemic has revealed that this was a tragic mistake. Potent antivirals, like those discovered many years ago for the related SARS and MERS coronaviruses, were never developed for clinical use. Millions of lives could have been saved if these compounds had been developed into drugs.
Direct-acting antivirals (DAAs) are defined as those that specifically inhibit viral enzymes that are needed for virus replication. Examples include the herpes drug acyclovir, AZT, the first drug approved to treat AIDS, and the hepatitis C drug sofosbuvir. The term was coined to differentiate DAAs from other antiviral drugs, like interferon and ribavirin, which function mainly by modulating the host immune system.
For over 20 years, the Frick lab worked with numerous other labs and companies to develop DAAs to treat hepatitis C. Many of these drugs are now used routinely in the clinic to cure chronic hepatitis C, which used to kill tens of thousands of Americans each year. With DAAs hepatitis C can now be cured with a single daily pill taken for eight weeks, with few if any side effects. The goal now is to adapt these DAAs for use with other viruses, like SARS-CoV-2, the virus that causes COVID-19.
Drug Target Identification
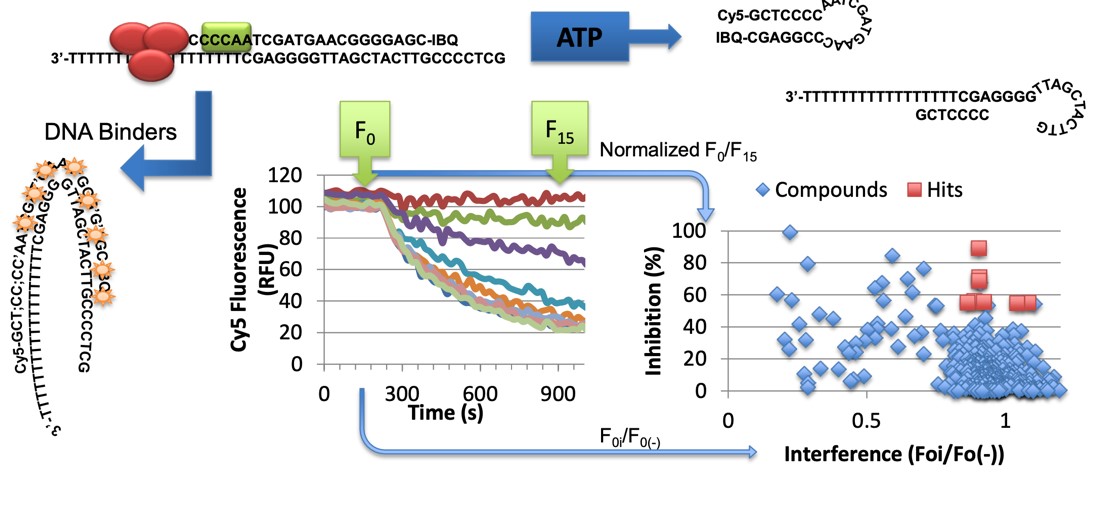
Most hepatitis C virus DAAs were discovered by biochemists using recombinant purified proteins because, unlike other viruses, HCV cannot be easily grown in the laboratory. To facilitate these efforts, scientists in the Frick lab, isolated viral proteins and designed assays suitable for high-throughput screening. These enzymes include viral polymerases, proteases, primases, and helicases. One example is an assay that can be used to discover viral helicase inhibitors and simultaneously differentiate them from toxic compounds that simply block viruses by binding nucleic acids (Fig. 1).
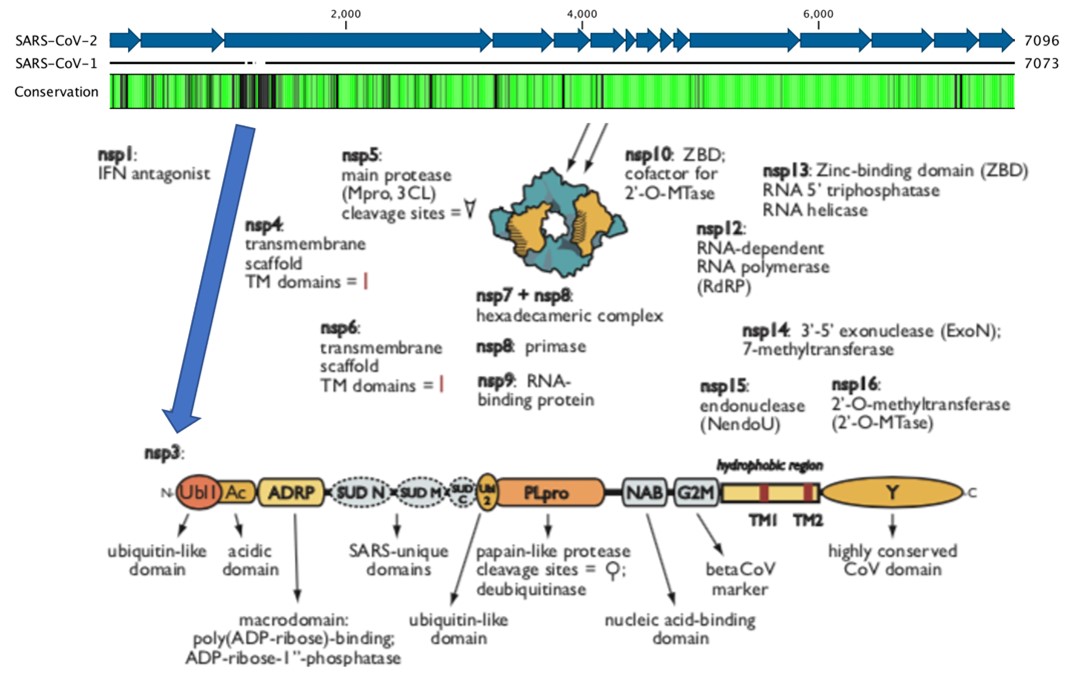
Because we desperately need DAAs to treat COVID-19, beginning in January 2020, the Frick Lab began isolating proteins from SARS-CoV-2. The 29,900 nucleotide SARS-CoV-2 RNA genome encodes many potential DAA targets, most of which are encoded by the rep1ab open-reading frame. The Frick lab has had most success with the multifunctional 945 amino-acid-long nsp3, which is tethered to the ER with two ubiquitin-like domains, two papain-like protease domains, three macrodomains (Mac1, Mac2, and Mac3), a nucleic-acid-binding domain, and a hypervariable region (Fig. 2).
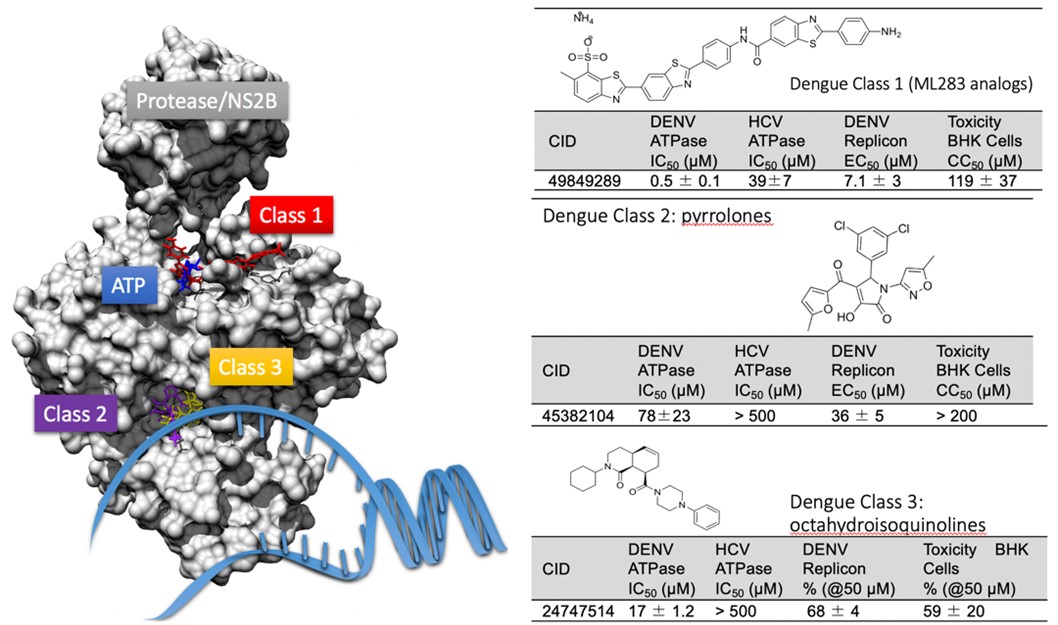
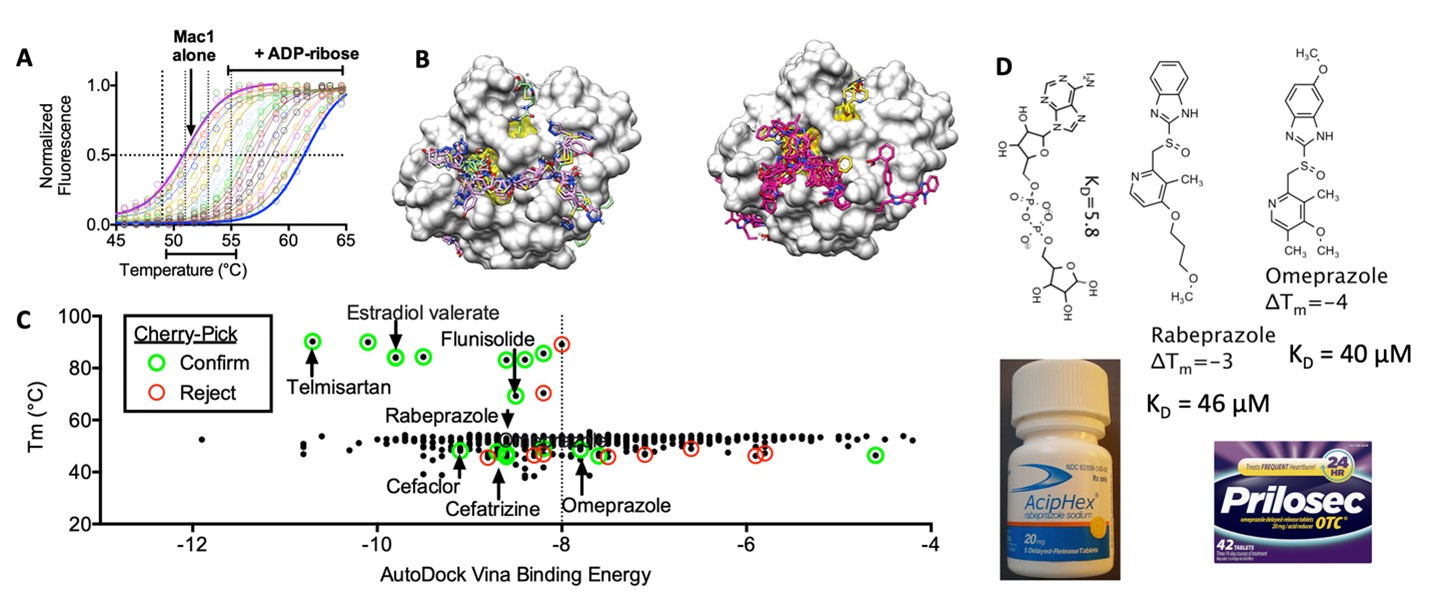
In Vitro Efficacy
After DAAs were approved and became the standard of care to treat hepatitis C, the Frick Lab began testing DAAs against similar viruses with RNA genomes like Dengue virus, West Nile virus, Zika virus and most recently SARS-CoV-2. Many were potent enzyme inhibitors and blocked virus replication (Figs. 3, 4). We now seek to develop this leads into antiviral drugs.